

Since the MDR was launched in May 2017, the companies of medical devices and in-vitro diagnostic medical devices who have been active on the European market made up their mind to keep up with the new trend. LAGIS has been one of the players in EU; therefore, getting the MDR certification became our commitment. As LAGIS is the first certified manufacturer of class IIa medical devices in Taiwan, in this article, we are going to share our case study in hope to provide useful references to our industry partners.
LAGIS divides the process of applying for MDR into four major parts: (1) pre-preparation (2) documentation submission (3) certificate issuance (4) implementation (5) post-market surveillance.
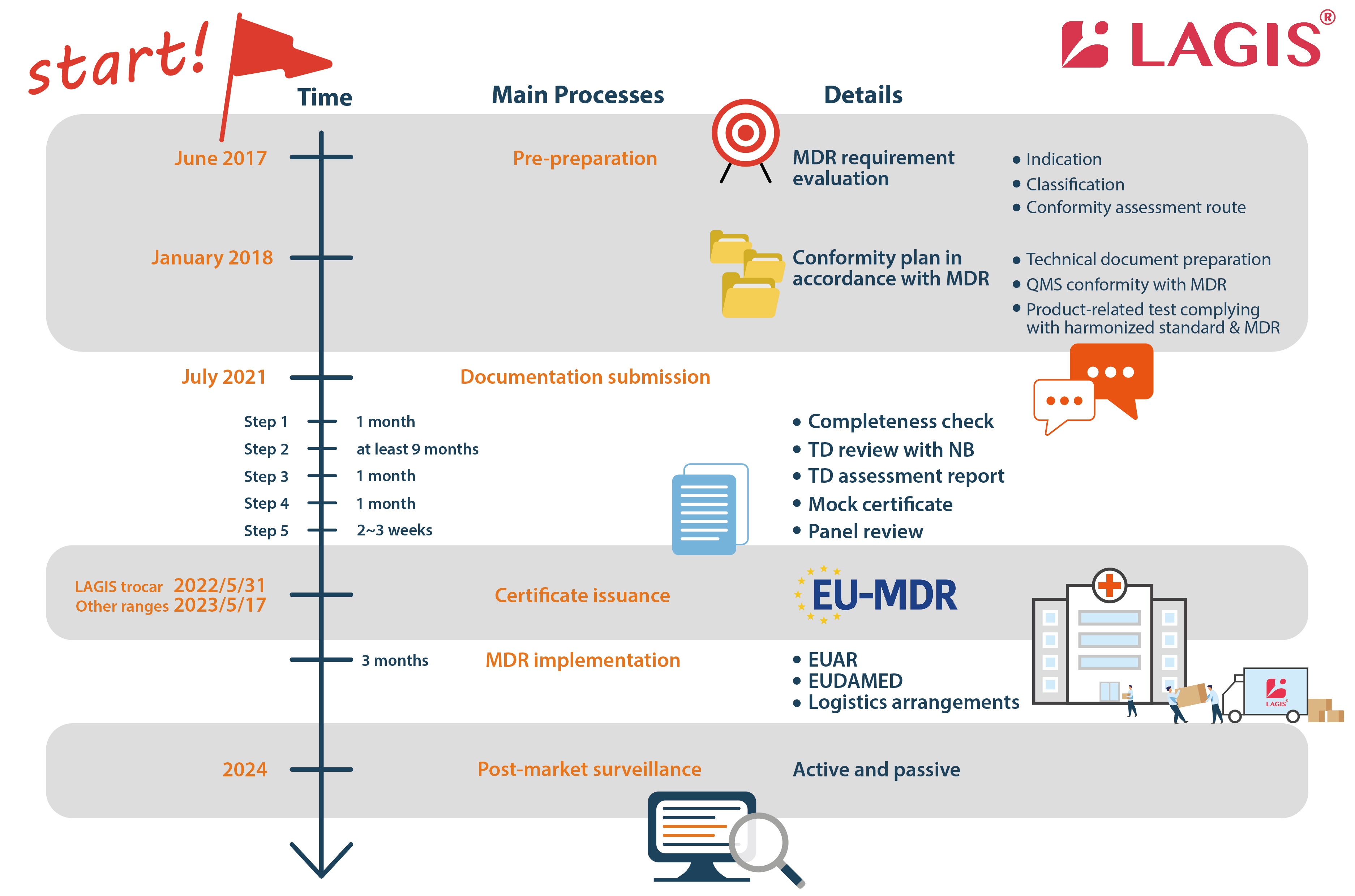
Pre-preparation
Once the MDR was announced, LAGIS set up a team with around 20 representatives from all departments (RA & QA, R&D, manufacturing, sales and administration) to drive the whole company to undertake the MDR transition plan. From 2018, we re-confirmed the products’ indication and classification and clarify which conformity assessment routes to follow. During the process, we carried out the gap analysis between regulations and in-house QMS to comply with the MDR (EU) 2017/745.
LAGIS officially submitted the MDR application to BSI on 14th July 2021 after the huge project of internal harmonization with the EU MDR regulation.
Documentation submission in five steps (around/at least 12 months in total)
1. Required document check (around 1 month)
Offer information according to the technical documentation required document check list provided by Notified Body.
If you would like to know the required document check list from our Notified Body BSI, please refer to the following link (page 11 to 38) to have more details: https://reurl.cc/N0NADm
2. TD review with NB (at least 9 months)
The review includes 3 main domains: TD file, microbiology, and clinical evaluation. The reviewer checks each domain and proposes questions progressively within 3 rounds. The most questions will be proposed in the first round. The less but the toughest questions will be asked in the third round. Each round takes at least 1 month. This is the most time and cost consuming stage because many reports should be re-conducted, and many rationales should be pointed out to prove the conformity of the medical devices.
As the MDR is more stringent than MDD, not only the validation of safety and performance is stricter, but also the post-market surveillance. Manufacturers should do their best to present the technical documentation in logical, quantified, and concrete ways to meet the requirements of Notified Body.
3. TD assessment report (around 1 month)
The Notified Body prepares the closing report.
4. Mock certificate (around 1 month)
After the mock certificate is provided, the manufacturer needs to confirm if the contents are correct.
5. Panel review (around 2-3 weeks)
Each medical device will be reviewed again by the committee which is composed of professionals from various fields. If there is any information to be modified or explained, the Notified Body will normally ask for supplementary documents instead of rejecting the application.
Certificate Issuance (around 2~3 weeks)
This is the most exciting result of the long conformity assessment procedures. The qualified medical devices can be placed on the market.
MDR implementation (around 3 months) and Post-market surveillance (continuously)
Once the certificate is on hand, it is necessary to inform the EUAR of this good news, then upload the data of the devices on the EUDAMED and each country’s national registration platform. The EUDAMED database provides information related to actor registration, unique device identifier (UDI), device registration, Notified Body & certificate, clinical investigation & performance studies, and vigilance & market surveillance. As the data is comprehensive and transparent, it will be useful for the market surveillance afterwards. For the logistics parts, the instruction for uses (IFU) and labels of the products are also renewed before the shipment to ensure that there will be no customs clearance problems.
LAGIS is pleased to have and to share such valuable experience. We hope that you get basic knowledge of the MDR application process. This opportunity will greatly benefit our team, making it a worthwhile investment. In the newsletter of August, we will invite our QA representatives in charge of MDR application to share their insights and experience on running this big project. If you want to have further information of LAGIS MDR products, please do not hesitate to contact our sales representatives: sales@lagis.com.tw

We use cookies to provide the services and features offered on our website, and to improve our user experience. By using this website, you consent to the use of cookies.