

In our previous articles, we provided a concise overview of the challenges and advantages associated with Medical Device Regulation 2017/745, along with an exploration of the primary application process and preparation. Now, we are delighted to present an opportunity for you to gain further insights and valuable firsthand experiences from our QA representatives responsible for the MDR application.

Was the announcement of MDR a sudden news to LAGIS?
The highest standard of regulation is to preserve the spirit of the state-of-the-art. The medical pioneers continually strive to enhance the quality of medical devices across product development, clinical evaluation, and regulatory aspects. They build up good models for the partners to follow, improve and advance together. LAGIS usually attends seminars to stay abreast of the latest revisions in medical regulations worldwide. Before the announcement of the MDR, there were sayings that the European Directive 93/42/EEC would be revised to prevent some scandals from happening again. When ISO 13485:2003 converted to ISO 13485:2016 version, the regulatory compliance was greatly emphasized. Additionally, during the era of MDD 93/42/EEC, certain guidance regarding clinical assessment, post-market surveillance and vigilance were complemented. This action allowed us to foresee the structural change would take place as a result. The evolution mentioned above can be considered the prelude to the birth of the MDR. Therefore, the publish of the new regulation (EU) 2017/745 was not a surprise. The biggest difference after its launch is that certain guidance that were originally non-mandatory have now become essential requirements. For example, in the clinical evaluation part, there is an exclusive Annex in the Medical Device Regulation (EU) 2017/745 where the related terms are also defined, and the overall requirements are stricter than before to verify the safety and performance of the medical devices.
Understanding the regulations should be a challenge, how do you get the staff to know the essence of MDR?
(As mentioned in the previous article, LAGIS established a cross-functional team to implement the MDR transition plan.) Actually, we have similar experience while transitioning from ISO 13485:2003 to ISO 13485:2016. The case study helped us to understand the keys of the transition process. After I (RA manager) read the Medical Device Regulation (EU) 2017/745 several times and completed the preliminary judgement, I gathered representatives from all the departments to study and summarize the articles together. (RA manager arranged the reading group meticulously.) Articles were assigned to relevant staff based on topics, making the regulatory requirements easier to digest and more practically tied to current workflow. Through the reflection and inspiration from key members, we could know how to adjust each in-plant task more comprehensively and objectively according to the MDR. After that, analysis results were also documented as training materials. We organized training courses for the middle managers, allowing them to lead the employees to implement the spirit of MDR into their work.
How to continually ensure the conformity with the MDR?
In terms of products, we established SOPs for post-market surveillance to ensure the safety and performance of products. When it comes to the in-plant process, we ensure the compliance with the regulations and guidance through regular QMS audits. We integrate the requirements of MDR and ISO 13485, two complementary requirements, into assessment checklist and this become one of the guides of our QMS audits. To preserve the "state-of-the-art" spirit, we regularly hold meetings to discuss the latest additions and updates in medical regulations and standards. During these meetings, we ensure to identify and take necessary actions to comply with any new guidance that should be followed.
Which is the most difficult application process of MDR?
Absolutely, it is the stage of technical document review with NB. And it is the core of the MDR conformity assessment procedures. LAGIS products have been on the market for near 20 years, so the quality, safety and performance have been recognized. The TD review is difficult because the regulations should be converted into documented SOPs and put them into practice, and any evidence to support compliance with GSPR (General Safety and Performance Requirements) should be provided. For instance, the design control matrix needs to present the verification / validation results or appropriate rationale for each design requirement, from user requirements to the forming of product. For products that have been on the market for many years, it took a longer time to sort out and trace relevant information and supporting evidence. Because of this, we reviewed the product development history, like reading a memoir, seeing the process of our efforts. Thanks for the valuable experience, we also optimized the design and development SOP and other workflows in accordance with the MDR.
From my point of view, the trickiest part is the TD review with NB, particularly, in two parts, the technical documentation and cross-departmental cooperation. The assessment level of Notified Bodies has intensified because the MDR became stricter than before, demanding technical documentation to cover more comprehensive aspects, including TD file, microbiology, and clinical evaluation. How to document the evidence in logical, quantified, and concrete ways is a challenge. For the latter one, As the project owner, my responsibility is to clarify the requests from the Notified Body in order to collect relevant information in collaboration with RD, to demonstrate the safety and performance of the devices. The challenge of this process lies in presenting the reply in a way that reach a balance between internal expertise and regulation requirements. The other tough but not difficult thing was the certificate issuance process. Although LAGIS products had passed the panel review, it still took a long time to get the final certificate due to the limited capacity of Notified Body. Originally, we were full of expectations to get the MDR certificate soon after the panel review, but the time for obtaining the certificate was delayed, and we were overwhelmed by pressure from inside and outside. To solve this problem, we worked hard to contact the scheme manager, and at the same time contacted the Notified Body in the United States and Taiwan and tried our best to track the progress of our case.
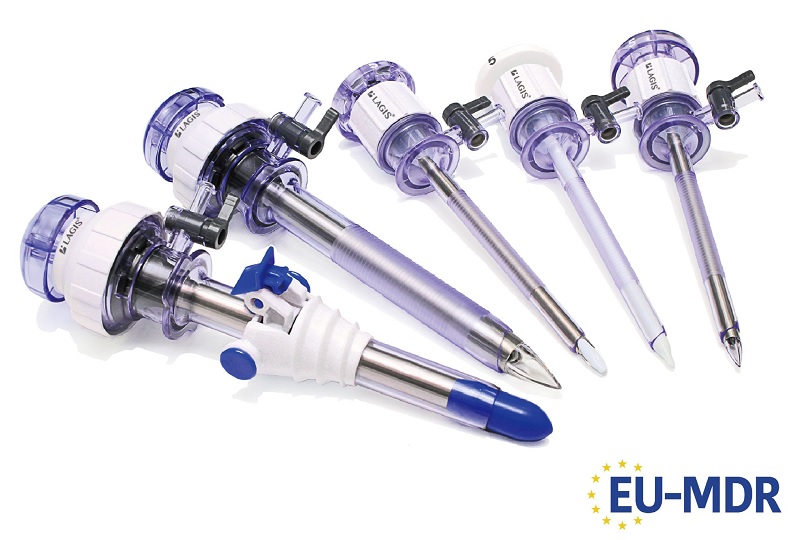
The CE certificate stands as a fundamental requirement for placing medical devices in numerous markets, spanning beyond the European Union to Asian and African countries like Hong Kong, Israel, South Africa, and more. Certain regions may request other international certificates such as FDA, MDSAP, or national certificates like ANVISA, TGA, PMDA, KFDA, and others. At LAGIS, we are committed to diligently pursue comprehensive registration to facilitate worldwide market development for our products. Please do not hesitate to contact LAGIS representatives for further information and cooperation.


We use cookies to provide the services and features offered on our website, and to improve our user experience. By using this website, you consent to the use of cookies.